
“Flowers of South-West Europe - a field guide” - de Oleg Polunin e B.E. Smythies
“Revisitas” de regiões esquecidas no tempo - “Plant Hunting Regions” - a partir de uma obra de grande valor para o especialista e amador de botânica como da Natureza em geral.
Por
Horst Engels, Cecilia Sousa, Luísa Diniz, Nicole Engels, José Saraiva, Victor Rito
da
Associação “Trilhos d’Esplendor”
2.13 The Northern Serras of Portugal
2.13 As Serras do Norte de Portugal
2.13.2 Serra da Estrela
|
Folha de Cálculo: Flora da Serra da Estrela
(Lista provisória de plantas vasculares e não-vasculares)
|
Bases de Dados:
|
Mapas das Serras do Norte de Portugal:
|
2.13.2 Serra da Estrela
2.13.2d3 - Aspectos glaciários - Filogeografia, Refúgios e Endemismos
Na nossa viagem no tempo chegamos à Serra da Estrela - e percebemos que o tempo não tem passado apenas para nós, mas que houve uma longa história evolutiva nesta região, passagem que mudou não apenas os aspectos paisagísticos desta serra (como vimos nos capítulos anteriores), mas em que deve ter havido também refúgios glaciares e recolonizações inter- e pós-glaciares por diversas espécies de plantas e animais (veja também nos artículos anteriores), já muito antes que o homem descobriu a Serra da Estrela para os seus fins agro-pastoris (há cerca de 7000 anos). Já mencionamos duas espécies, Cryptogramma crispa e Galemys pyrenaicus, que habitam a Serra da Estrela provavelmente deste à última glaciação (Würm) e que são espécies relíquias com refúgios pleistocénicos na Península Ibérica. Também a espécie Chionomys nivalis é uma espécie que teve refúgios glaciares do Pleistocénico na Península Ibérica, mas a sua história biogeográfica está desconhecida para a Serra da Estrela. Sem dúvida a Península Ibérica teve um papel importante como “refúgio mediterráneo” no Pleistocénico para espécies de plantas e animais que mais tarde (re)-colonizaram a Europa Central. Mas além disso possui a Península Ibérica a sua própria história filogeográfica e biogeográfica, - além de ser provavelmente um importante núcleo evolutivo de espécies.
No entanto, a biogeografia como ciência histórica parece ser necessariamente especulativa. Até há pouco esta disciplina baseou-se principalmente no estudo morfológico de espécies e de fósseis. Umas decadas atrás, com a introdução de métodos bioquímicos na genética populacional, começavam a ser usados também genes (alleles) nesta disciplina.
Ao mesmo tempo foram desenvolvidos métodos comparativos na sistemática e biogeografia, baseado nos estudos e publicações de Willi Hennig (“Grundzüge einer Theorie der phylogenetischen Systematik” (publicao em alemão em 1950 - traduzida para inglês em 1966 (“Phylogenetic Systematicas”) e de Leon Croizat (1964 - “Space, Time and Form: The Biological Synthesis”) que permitiam usar espécies recentes (actuais) na reconstrução filogenética e que revolucionaram em pouco tempo a sistemática e biogeografia. Os métodos cladísticos e comparativos[1][2] são usados sobretudo em espécies recentes (sem depender de fósseis) para a reconstrução filogenética e filogeográfica. (Um bom livro que trata os aspectos cladísticos e comparativos na Sistemática e Biografia é o livro: “Systematics and Biogeography. Cladistics and Vicariance” de Gareth Nelson and Norman Platnick (1981).)
Pensava-se até há pouco que os métodos cladísticos e comparativos apenas se aplicavam em linhas filogenéticas acima do nível da espécie, uma vez que as ramificações filogenéticas das espécies têm correspondência numa hierarquia de caracteres taxonómicos que podem ser agrupados (“nested characters”) e cujo poder discriminativo em linhas filogenéticas diferentes tem causas nas barreiras genéticas que surgem e acompanham o processo evolutivo das espécies. O método comparativo não parecia aplicável aos níveis taxonómicos inferiores da espécie e uma solução para o estudo da evolução filogenética infraespecífica parecia por muito tempo inatingível e intractável. Mas esta visão estava no mínimo incompleta. John C. Avise foi um dos pioneiros à mostrar (ha mais de 20 anos) que a genética molecular e métodos comparativos podem bem ser usados para a reconstrução de árvores genéticas numa linha filogenética de populações de espécies usando a informação da demografia histórica e da genealogia populacional das mesmas. Através da determinação do “coalescent”, ancestral comum mais recente numa linha genealógica (populacional) de uma espécie, pode ser reconstruido a filogeográfica da espécie. Foi a partir daí que um novo ramo científico começou a crescer. A teoria de coalescência (uma teoria matemática e estatística) foi desenvolvida (a partir dos anos 80) servindo hoje de fundamento estatístico para a filogeografia[3][4]. Um resumo sobre a filogeografia estatística encontra-se no artigo: “Statistical Phylogeography” de L. Lacey Knowles[5][6].
Existem neste momento 3 abordagens principais na filogeografia: (1) o procedimento clássico e comparativo (comparative approach) em que é usado normalmente o método “Nested clade phylogeographical analysis (NCPA)”[7][8] de Alan R. Templeton, (2) um método de difusão espacial (Spatial diffusion approach), e (3) os métodos da genética populacional (Population genetics approach) na base do “coalescent estruturado”.[9][10]
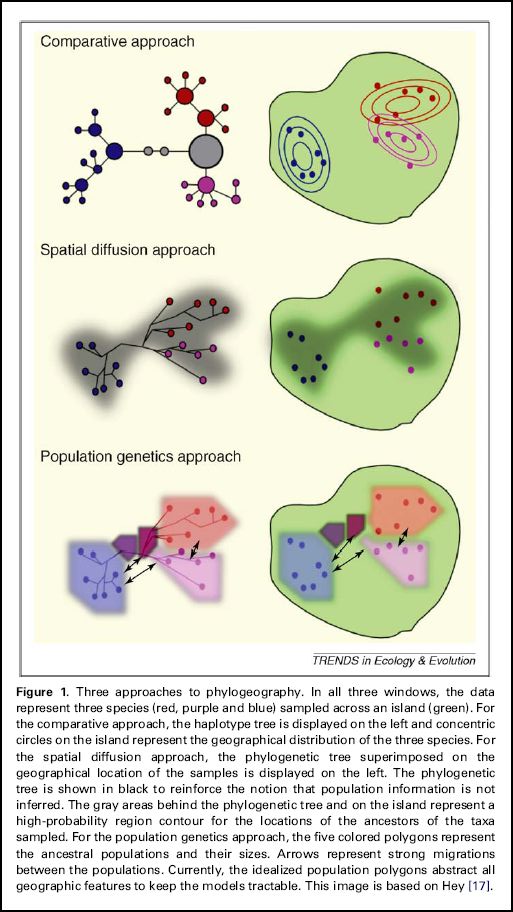
Trends in Ecology and Evolution Vol.25 No.11
Outro resultado da investigação actual na filogeografia é a compatibilização e integração da teoria de coalescência (desenvolvido à partir da teoria neutralísta da evolução para populações de espécies no âmbito da genética populacional) com a teoria filogenética clássica de seleção natural - quer dizer integrar “gene trees” de populações de espécies na reconstrução de “species trees” que serviram e servem de base para uma sistemática filogenética de Willi Hennig e de outros cientistas da sua epoca.[11][12]
A filogeografia tira assim não apenas partido do paradigma da “teoria neutralísta da evolução” - desenvolvida por Motoo Kimura e apresentada no livro “The Neutral Theory of Molecular Evolution”, mas as metodologias estatísticas, por vezes muito complexes, tornam a filogeografia numa ciência testável e falsificável (no sentido de K. Popper) - que já não necessita basear-se apenas em narrativas. Actualmente, filogeografía combina diversos instrumentos científicos sofisticados para a reconstrução da biogeográfica (numa dimensão espacio-temporal) das espécies.
Também o ramo da antropologia tira parte destas novas tecnologias na reconstrução filogeográfica para a reconstrução do caminho evolutivo da espécie humana (incluindo as migrações do homem no passado pré-histórico).[13]
Um bom enquadramento com retrospectiva e prospectiva da filogeografia que transmite também as ideias básicas desta disciplina encontra-se no artigo “Phylogeography: retrospect e prospect” de John C. Avise (2009)[14]:
ABSTRACT
Phylogeography has grown explosively in the two decades since the word was coined and the discipline was outlined in 1987. Here I summarize the many achievements and novel perspectives that phylogeography has brought to population genetics, phylogenetic biology and biogeography. I also address future directions for the field. From the introduction of mitochondrial DNA assays in the late 1970s, to the key distinction between gene trees and species phylogenies, to the ongoing era of multi-locus coalescent theory, phylogeographic perspectives have consistently challenged conventional genetic and evolutionary paradigms, and they have forged empirical and conceptual bridges between the formerly separate disciplines of population genetics (microevolutionary analysis) and phylogenetic biology (in macroevolution).
|
INTRODUCTION
Phylogeography is a relatively new discipline that deals with the spatial arrangements of genetic lineages, especially within and among closely related species. The word phylogeography was coined in 1987 (Avise et al., 1987). About a decade earlier, scientists had begun using mitochondrial (mt) DNA to address precisely how conspecific individuals are genealogically linked through shared ancestors (the mitochondrial genome is well suited to this purpose, for reasons that this paper will highlight). In genetic surveys of various species, striking patterns were being uncovered in the spatial arrangements of mtDNA lineages. In other words, genealogy and geography seemed to be connected. After 1987, ‘phylogeography’ became the convenient shorthand for referring to explicit genealogical inquiries (including those for nuclear genes) into the spatial and temporal dimensions of microevolution.
In physics, Albert Einstein’s Theory of General Relativity formalized the notion that time and space are interrelated and that the curvature of space–time predictably affects the paths of free particles. In phylogeography, no comparable mathematical laws govern precisely how genetic lineages are spatially configured (evolutionary processes are much too varied and idiosyncratic for that). However, comparative phylogeographic assessments of many species have revealed a great deal about the demographic and historical nature of intraspecific evolution.
I begin this overview with some by-now-standard background on cytoplasmic genomic data and on coalescent theory (Hudson, 1990) which are often used in phylogeographic appraisals. Then I will illustrate, by empirical examples, some key findings and emerging principles of phylogeography, before concluding with some thoughts about future directions for this burgeoning discipline. This paper offers an introductory synopsis of the field; for a more in-depth treatment and extensive citations to the primary phylogeographic literature, see Avise (2000).
MOLECULES AND ASSAYS
Mitochondrial DNA evolves rapidly in animals
Mitochondrial DNA in eukaryotes ultimately traces back to a proteobacterium that merged endosymbiotically with a pre-eukaryotic cell early in the history of life. Over evolutionary time, this cytoplasmic genome lost most of its DNA to the nucleus and today is typically represented in animal cells by only 37 genes all linked along a closed circular molecule about 17,000 nucleotide pairs in length. Two of these genes encode ribosomal RNAs, 22 specify different transfer RNA molecules, and 13 encode protein subunits that collaborate with nuclear-encoded polypeptides in the biochemical pathways of electron transport and oxidative phosphorylation that help generate cellular energy. The molecule also includes a control region (CR) where mtDNA replication is initiated. MtDNA replication is asynchronous with cell division, and is frequent, such that hundreds or thousands of copies of mtDNA normally inhabit the cytoplasm of each somatic or germline cell of an individual.
The mitochondrial genome of animals is highly compact; that is, it is not burdened with the introns or long intergenic stretches of non-coding spacer sequence that characterize nuclear genomes. Such genetic streamlining must be strongly selected for in the competitive cytoplasmic environment, where smaller mtDNA molecules probably replicate faster than larger ones and thereby gain a significant transmission advantage (all else being equal) from one cell generation to the next.
Given the important functional role of mtDNA in animal cells and the general structural economy of the molecule, pre- 1980s speculation had been that this genome should evolve slowly in nucleotide sequence. Surprisingly, this did not prove to be the case: as first shown by Wes Brown et al. (1979), animal mtDNA accumulates nucleotide substitutions several fold faster than does typical single-copy nuclear DNA. Several factors probably contribute to the rapid pace of mtDNA evolution: relatively inefficient mechanisms of DNA repair in mitochondria; the corrosive oxygen-rich environment to which mtDNA molecules are exposed in the mitochondrial organelle; a relaxation of functional constraints resulting from the fact that mtDNA encodes only a few types of polypeptides and does not produce proteins directly involved in its own replication, transcription, or translation; and the fact that mtDNA is naked (i.e. not tightly bound to histone proteins that themselves are conservative and may constrain evolutionary rates in nuclear DNA). The rapid evolution of mtDNA is also reflected in the presence, within most animal species, of high nucleotide sequence variation – a prerequisite for phylogeographic analyses.
Populations of mtDNA molecules exist within somatic and germline cells
Large populations of mtDNA molecules (often thousands or more) typically occupy each cell, thus adding a potential complication to genetic analysis that does not exist for nuclear genes. (Each autosomal gene, in contrast, occurs in only two copies per diploid cell and one copy per gamete.) However, a key empirical finding is that most individuals are homoplasmic for mtDNA, meaning that all of their mtDNA molecules are essentially identical in nucleotide sequence. Heteroplasmic animals (those showing within-individual mtDNA sequence variation) are detected occasionally, but in such cases the molecules typically differ by merely one or a few recently arisen mutations.
Why homoplasmy predominates is unclear, but one likely possibility is that mtDNA molecules in germline cells undergo relative population bottlenecks such that the large number of molecules in each zygote stems from far fewer precursor molecules in pre-meiotic germ-cell generations. If so, each genetic transition from heteroplasmy to homoplasmy, or from one homoplasmic state to another, could occur by genetic drift in relatively few animal generations (as seems to be true experimentally). Other theoretical possibilities are that natural selection favours homoplasmy, or that gene conversion mechanisms periodically homogenize mtDNA sequences in a cell lineage. Regardless of the underlying causes, homoplasmy is important for phylogeographic studies because each individual animal typically carries a single, specifiable mitochondrial genotype.
Maternal inheritance characterizes animal mtDNA
Another distinctive property of mtDNA is the maternal transmission mode of the molecule. During the fertilization process (syngamy), most of a zygote’s cytoplasm comes from the egg. Each offspring thus receives mtDNA primarily, if not exclusively, from its dam rather than from its sire.
Female transmission of mtDNA is analogous to male transmission of surnames. In many human societies, sons and daughters both assume their father’s surname, which only sons pass to their children; analogously, sons and daughters inherit their mother’s mitochondrial genotype, which only daughters pass to progeny. Several key differences do exist, however: surname transmission is a recent social convention (first used in China during the Han Dynasty, at about the time of Christ), whereas mtDNA transmission has gone on for hundreds of millions of years; surnames are unique to humans, whereas mtDNA ‘family names’ occur naturally in all animal species; and surnames seldom are more than about 12 letters long, whereas each mtDNA family name consists of about 17,000 nucleotide pairs (although only a subset of these are assayed in a typical molecular study). Another distinction is that a particular human surname may have many independent origins and thus be polyphyletic. This is because the source of many surnames is a common profession (such as Baker or Smith), a prominent historical figure (such as Williams), or conversion from an earlier system of patronymics in which a suffix was added to a person’s given name (yielding Johnson or Jackson, for example). By contrast, each distinctive array of mtDNA genotypes is almost certainly monophyletic.
Because mtDNA is uniparentally transmitted, molecules from different families seldom if ever recombine. In effect, mtDNA inheritance is both haploid and asexual. Because mtDNA lacks the shuffling aspects of meiosis and syngamy
that affect nuclear DNA during sexual reproduction, mutations alone normally account for the genetic variety of mtDNA often observed in animal populations. MtDNA genotypes are referred to as haplotypes, which differ from one another by particular mutations accumulated since female ancestors were last shared. Because of the rapid pace of mtDNA evolution, many different haplotypes typically coexist within a species.
Scientists can use these haplotype sequences to estimate the matrilineal histories of individuals and populations.
Organelle genomes play by different evolutionary
rules in plants
Plants have two cytoplasmic genomes – mtDNA and chloroplast (cp) DNA – which are likewise of endosymbiotic origin and have lost various genes to the eukaryotic cell nucleus over evolutionary time. Today, these organelle genomes are similar to animal mtDNA in overall structure (closed-circles), replication mode (yielding large populations of molecules per cell), and non-Mendelian inheritance patterns. However, as first emphasized by Palmer (1990), they also differ from animal mtDNA, and from one another, in some important molecular and evolutionary properties.
Plant mtDNA is highly variable in size, ranging from about 20 kilobases (kb) to 2,500 kb across species. Inheritance is often maternal, but not invariably so. Surprisingly, plant mtDNA evolves rapidly with respect to gene order (i.e. gene rearrangements are common) but slowly with respect to primary nucleotide sequence. Thus, in these regards the evolutionary dynamics of plant mtDNA and animal mtDNA differ diametrically. The slow pace of mtDNA sequence evolution in plants (about 100· slower than in animals) plus other technical complications mean that plant mtDNA is rather poorly suited for phylogeographic surveys within species.
Plant cpDNA plays by another set of evolutionary rules. It varies moderately in size among species (from about 120 to 217 kb in photosynthetic land plants, for example), with much
of the size variation attributable to the extent of sequence reiteration in a large inverted repeat region. The molecule contains about 120 genes that code for ribosomal and transfer RNAs plus numerous polypeptides involved in protein synthesis and photosynthesis. CpDNA is transmitted maternally in most species, biparentally in some, and paternally in others (notably, most gymnosperms). The molecule tends to evolve at a leisurely pace with regard to gene rearrangements and also in terms of primary nucleotide sequences (about 3· to 4· faster than plant mtDNA, but still much slower than animal mtDNA). For this latter reason, cpDNA sequences have proved especially useful for estimating phylogenetic relationships among higher taxa of plants. With diligent sequencing efforts, however, sufficient genetic variation can be uncovered for phylogeographic appraisals within particular species as well.
Plant cpDNA typically occurs in many copies in a cell’s cytoplasm. Nonetheless, most individuals display a single cpDNA haplotype sequence (and thus are homoplasmic), and various individuals within a species sometimes show detectable differences in cpDNA sequence. In these respects, plant cpDNA is much like animal mtDNA, and scientists can likewise capitalize on the molecule’s haploid, non-recombining nature to estimate genealogical histories in plant populations.
These histories may be matrilineal, patrilineal, or an unspecified mixture of the two, depending on the mode of genetic transmission of cpDNA in the species being investigated.
Nuclear loci also have genealogical histories
In principle, single-copy nuclear (scn) genes could also provide voluminous sequence data for phylogeographic assessments at the intraspecific level, but three technical and biological hurdles have impeded progress: the relatively slow pace of sequence evolution at many nuclear loci; the difficulty of isolating nuclear haplotypes, one at a time, from diploid organisms; and the phenomenon of intragenic recombination.
The first complication can be overcome by identifying and monitoring scn sequences that evolve more rapidly than average. For example, intron sequences in protein-coding genes often evolve faster than their adjoining exons. The second hurdle can be overcome by using natural or artificial systems of haplotype isolation. For example, genes on the X- or Y-chromosomes of mammals (or the Z- and W-chromosomes of birds) are present in haploid (hemizygous in this case) condition in each member of the heterogametic sex, as are all scn loci in males of species such as bees or ants with haplodiploid sex determination. When genes in haploid condition are not readily provided by nature (as is typically the case in diploid organisms), it is also possible to purify nuclear haplotypes from any species using laborious molecular methods (such as artificial cloning into biological vectors). Regardless of the method employed, for genealogical purpose it is important to assay haplotypes individually, because otherwise the phase (cis vs. trans) of particular haplotypes may remain unspecified in diploid specimens that prove to be heterozygous at multiple nucleotide sites along a gene.
The third hurdle merits elaboration because it is undoubtedly the most serious. In any population of sexually reproducing organisms, meiosis and syngamy continually re-assort unlinked genes into new multi-locus combinations. These recombinational processes can also manifest within a locus whenever successful meiotic crossover events occur between different alleles. Each recombinant allele that emerges is then an amalgamated stretch of DNA, the subsets of which might have quite different evolutionary pasts. Thus, intragenic (or inter-allelic) recombination tends to complicate and garble what would otherwise be cleaner genealogical signatures from the nuclear genome. This contrasts with the historical clarity characteristic of the ‘blessedly celibate’ haplotypes in nonrecombining cytoplasmic genomes (Dawkins, 1995).
Nonetheless, scnDNA haplotypes have been employed successfully in various phylogeographic assessments (Hare, 2001), with some of the most informative results coming from intron sequences at protein-coding genes. Furthermore, even when the technical and biological hurdles of extracting explicit genealogical data from scnDNA sequences prove insurmountable, less direct information about population histories can still be obtained from conventional population genetic analyses that focus on the observed frequencies of phylogenetically unordered alleles. Assays of allozymes and microsatellites, for example, are used routinely to estimate population allele frequencies that in turn can be used in spatial genetic analyses of conspecific populations. Especially when such data are accumulated across multiple unlinked loci, historical information often emerges that can be compared with the more explicit genealogical patterns that normally are apparent in cytoplasmic genomes.
COALESCENT CONCEPTS
All members of a population are genealogically linked
Although many distinct genetic lineages (analogous to family names) may currently be present in a population, they all trace back to particular shared ancestors at some point in history. To grasp this concept, consider first the matrilineal history of extant individuals in any given species. If the true population pedigree were known, any genealogist could trace the female lineages back through time, and eventually reach a great-great-great-great…grandmother, some number of generations earlier, that was shared by all individuals alive today. Thus, all extant individuals would track back, ultimately, to this one individual via a coalescent process involving matrilines. Consider next the extant population’s patrilineal history. By tracing back through male lines, eventually a shared great-great-great-great…grandfather would likewise be reached through a comparable coalescent process. An important point is that the matrilineal and patrilineal genealogies are each tree-like (branched and non-reticulate) despite being embedded in an organismal pedigree that overall is highly anastomose by virtue of interbreeding. Each such branching diagram (i.e. the collection of pathways that is strictly matrilineal or partilineal) can be thought of as a gene tree or gene genealogy within an organismal pedigree.
A distinction should be made between idealized gene genealogies on the one hand and imperfect empirical estimates of gene trees on the other. A quintessential matrilinea genealogy, for example, is by definition the actual history o genetic transmission from female to female to female across the generations, and a quintessential patrilineal tree is the actual history of genetic transmission from male to male to male. Such genealogical histories are never known directly for species in nature, but their principal features can be estimated from suitable DNA sequence comparisons of living animals.
Cytoplasmic genomes in particular can provide information on matrilineal histories, and the mammalian Y-chromosome can provide information on patrilineal histories.
Groves of gene trees grow in any population or species
At first glance it might seem that the sum of matrilineal and patrilineal histories would complete each picture of genetic ancestry. However, this is far from true in any population of sexually reproducing organisms, because most nuclear genes are transmitted via both genders. Consider, for example, the fact that any individual in a non-inbred pedigree had eight great-grandparents from each of whom it inherited approximately one-eighth of its autosomal DNA, but that only two of those great-grandparents (25%) were that individual’s matrilineal and patrilineal ancestors.
Conceptually, any organismal pedigree can be decomposed into multitudinous gene genealogies (Fig. 1) that are analogous to the matrilineal and patrilineal trees described above, but which involve transmission histories through male (M) and female (F) ancestors (as would be true for any nuclear autosomal locus). For example, one such hypothetical pathway might involve alternate genders (F, M, F, M, F, M, F…) across the preceding generations; another could be M, M, F, F, M, M, F, F…. In any pedigree that is G generations long, the theoretical number of distinct ‘gene trees’ of this sort (as defined by gender-specified genealogies) is simply 2G.
In the real world, no autosomal gene is transmitted via females only or via males only, but instead both genders are involved (albeit not in a precisely specifiable gender-based pattern across multiple generations). In any event, in each generation the number of transmission pathways available for alleles at any particular autosomal gene is in effect fourfold greater (M fi F, F fi M, M fi M and F fi F) than that for mtDNA (F fi F only) or the Y-chromosome (M fi M only). This also implies that the mean coalescent time for an autosomal gene is expected to be about fourfold more ancient, all else being equal, than that for mtDNA or the Y-chromosome. This baseline fourfold difference can also be seen as stemming from a twofold effect resulting from diploidy for autosomal genes (vs. haploidy for mtDNA) coupled with another twofold effect resulting from transmission through two sexes (vs. only one sex for mtDNA or the Y-chromosome).
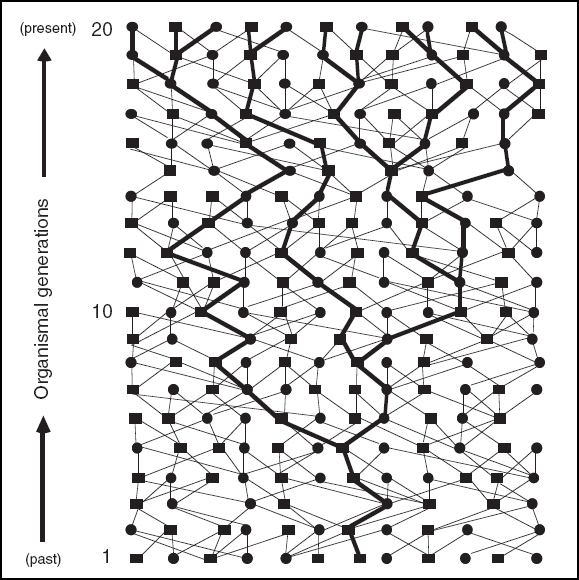
Figure 1 Hypothetical gene tree or haplotype tree (heavy lines) within a 20-generation organismal pedigree. Squares are males, circles are females, and lines connect parents to their offspring. The concepts of gene trees and coalescent processes have been central to the emergence of phylogeography as a recognizable subdiscipline of biogeography.
The broader point is that multitudinous distinct nuclear gene genealogies co-exist within the pedigree of any sexually reproducing population. In other words, unlinked segments of DNA have quasi-independent transmission histories such that their genealogical structures inherently differ, at least to some degree, from locus to locus.
Genealogy and historical demography are intertwined
Lineage sorting inevitably accompanies organismal reproduction and produces the kinds of gene genealogies discussed above. With respect to matrilines, for example, lineage sorting would fail to take place only if each female in each generation replaced herself with exactly one daughter, in which case there would be no hierarchical branching of matrilines and no coalescence. But in any real population, different females contribute unequally to the total pool of progeny, so each matrilineal tree inevitably self-prunes as some branchesproliferate and others die off. Differential reproduction thus underlies the inevitable coalescent process.
To illustrate, imagine that females produce daughters according to a statistically tractable Poisson distribution with mean (and variance) equal to 1.0. The Poisson assumes that successive daughters from a mother are independent and random events. Under this model, the expectation that a female contributes no daughters to the next generation (or, equivalently, the expected frequency of daughterless mothers) is e)1 = 0.368 (e is the base of the natural logarithms), and her probabilities of producing n offspring are given by e)1(1/n!). Thus, the probabilities of 1, 2, 3, 4 and 5 or more daughters are 0.368, 0.184, 0.061, 0.015 and 0.004, respectively. These values apply across one generation, but the calculations (which become more complex) can be extended to multiple generations. They show, for example, that about 98% of all founding matrilines are likely to go extinct within 100 generations, even when population size remains stable, because the other 2% of matrilines have survived and proliferated.
The broad conclusion from such statistical theory is that historical demographic factors (such as means and variances in offspring numbers per family) can impact gene genealogies in statistically predictable ways. For example, coalescent points in gene genealogies tend to be more ancient in populations with larger effective sizes, all else being equal. Thus, empirical data on the depths and branching structures of gene trees (such as estimated from mtDNA) can be interpreted under coalescent theory to help reveal the demographic history of a population.
Some of the insights from coalescent theory tend to be quite counterintuitive. For example, coalescence to a single ancestor in a matrilineal genealogy does not imply that only one female was alive in that coalescent generation; it merely indicates that other females in the coalescent generation were not survived to the present day by matrilineal descendants. Another important point is that coalescence in a matrilineal genealogy does not imply that other females in the ancestral population failed to contribute genetic material to later generations. Nuclear genes from many of those other individuals may well be represented in descendants by alleles that percolated through non-matrilineal pathways of the pedigree. Analogous statements and concepts likewise apply to patrilineal and all other genealogical pathways within an extended organismal pedigree.
Spatially structured populations are genealogically linked
Phylogeographic studies normally deal with multiple populations
distributed across a landscape. Behavioural and/or physical impediments to dispersal often exist between some of those populations, now or in the past. Especially in species with limited vagility (movement capability), populations may be partially isolated by distance alone. Physical filters or barriers (such as rivers for terrestrial species, or mountains for lowland species) often augment the effects of limited vagility in promoting population structure. Some such filters may be semi-permeable and permit occasional exchanges of genetic lineages, but others may block all gene exchange for substantial periods of evolutionary time. In the latter case, the overall coalescent point for any gene genealogy can be no less old than the vicariant (splitting) barrier itself. The goal in phylogeographic appraisals is to use gene trees to infer the historical and contemporary forces that have produced the current genealogical architecture of populations and closely related species.
For any species that has been genetically sundered by a firm dispersal impediment, genealogical patterns depend in principle on the barrier’s duration as well as on the historical demographies of the disjunct populations. Consider, for example, a random mating ancestral population that becomes sundered into two separate populations (A and B) by a vicariant event. Initially, it is quite likely that populations A and B would appear polyphyletic with respect to maternal ancestry (or mtDNA relationships), meaning that some individuals in A are truly genealogically closer to some individuals in B than they are to other individuals in A (and vice versa). After the passage of some number of generations, however, lineage sorting would probably yield an outcome in which population A has become paraphyletic with respect to B (or perhaps vice versa), meaning that some individuals in A are genealogically closer to some individuals in B than they are to other individuals in A but all individuals in B constitute a monophyletic unit within the combined genealogy of populations A plus B. After even more generations have passed, A and B would eventually become, via further lineage sorting, reciprocally monophyletic with respect to matrilineal ancestry, meaning that all individuals within each respective population are one another’s closest genealogical kin for that specified component of the pedigree.
This genealogical transition from polyphyly to paraphyly to reciprocal monophyly in a gene tree is an expectation of coalescent theory whenever a well-mixed ancestral population becomes sundered into isolated units (the polyphyly phase may be bypassed if the ancestral population was already subdivided prior to the barrier’s emergence). Temporal durations of the transitional genealogical stages also depend on the sizes of the daughter populations, because the entire genealogical process is reflective of lineage sorting, or of lineage turnover owing to differential reproduction. The important concept here is that genealogical patterns themselves can be evolutionarily dynamic (rather than static) following a population splitting event.
Genealogical concordances are evidence of deep phylogeographic structure
Given the expected heterogeneity of genealogical pathways within any species, and the multifarious behavioural, ecological, and evolutionary forces that can shape gene trees, how can the phylogeographic histories of populations be reconstructed from molecular data with any great confidence? Specifically, how can historically deep (ancient) population genetic subdivisions be empirically distinguished from those that are evolutionarily shallow (recent)? Answers to these questions are found by reference to principles of genealogical concordance (Avise & Ball, 1990).
In principle, at least four distinct aspects of genealogical concordance or agreement across characters can be recognized.
Aspect I concordance is a within-locus concept: if multiple nucleotide substitutions along a DNA sequence consistently distinguish one array of haplotypes from another, a deep (as opposed to shallow) historical split is strongly implied for branches in the gene tree under investigation. This is true because the accumulation of new mutations is a rather slow process, so appreciable time since coalescence must have elapsed whenever particular non-recombining DNA haplotypes show large sequence differences. If a genealogical split is documented merely in one gene tree, a longstanding historical split at the population level is not necessarily registered, however, because deep splits in particular gene trees can occur for other reasons as well (such as balancing selection, which can favour the long-term retention of highly distinct alleles at a locus).
Aspect II concordance is a multi-locus concept: if deep splits in multiple gene trees consistently distinguish the same sets of populations, the responsible evolutionary forces must have had pervasive genomic impacts. The most likely candidate for such pervasive effects across loci is a longstanding cessation of gene flow, such as would arise from lasting physical or behavioural impediments to dispersal. Thus, any empirical documentation of aspect II concordance strongly implies a long-term historical separation at the population level.
Aspect III concordance is a multi-species concept: if spatially consistent genealogical splits appear across multiple codistributed species, the responsible evolutionary forces must have had widespread effects at the level of biotic communitiesor ecosystems. Finally, aspect IV concordance refers to any agreement among multiple lines of empirical evidence. For example, if a pronounced gene-tree split in a particular species is geographically consistent with a longstanding dispersal obstruction that is known or suspected from independent evidence (e.g. morphology, subspecies taxonomy or physical geology), a deep phylogeographic split within the species is again strongly implicated.
Some of the coalescent concepts discussed in this section may seem esoteric. However, they all have real-world implications for interpreting empirical phylogeographic patterns, as the next section will demonstrate.
EMPIRICAL PHYLOGEOGRAPHIC PATTERNS
Strong genealogical structure characterizes low-dispersal species
The first substantial phylogeographic appraisal of mtDNA involved the south-eastern pocket gopher, a rather sedentary fossorial rodent (Avise et al., 1979). Among 87 animals genetically surveyed throughout the species’ range in Georgia, Alabama and Florida, more than 20 distinct mtDNA haplotypes were detected. Some of these differed by mutations at only one or two surveyed nucleotide positions, whereas others were much more divergent. All of the mtDNA haplotypes proved to be spatially localized, and closely related haplotypes were normally geographically proximate. In addition, a deep genealogical split distinguished all gophers in eastern vs. western portions of the species’ range. The shallow splits in the gene tree may evidence routine isolation by distance, whereas the deep split (estimated 3% sequence divergence) probably registers a much longer history of genetic separation – perhaps more than a million years since common female ancestry. This early empirical study in effect marked the birth of phylogeography as a recognizable discipline.
In the last 30 years, similar phylogeographic appraisals based on mtDNA sequences have been conducted on literally hundreds of low-vagility animal species ranging from various small mammals, reptiles and amphibians to invertebrates such as land snails and crickets. For such dispersal-challenged organisms, phylogeographic outcomes are typically like those described above: within each species, closely similar or identical mtDNA haplotypes are geographically localized, and a small number (typically one to six) of deeper genealogical splits often distinguish regional sets of populations that probably differentiated in separate refugia (Avise, 2000). The deeper splits often tend to align with known or suspected historical impediments to dispersal, such as the Isthmus of Panama, which rose above the sea about 3 Ma and vicariantly sundered many tropical marine species (fishes, sea urchins, etc.) into Atlantic and Pacific populations that today are genealogically quite distinct.
Comparable phylogeographic appraisals, based on cpDNA, have been conducted on several plant species. For example, among 85 populations of the common beech tree that were genetically surveyed across the species’ range in continental Europe, 11 distinct cpDNA haplotypes were detected (Demesure et al., 1996). Some of these haplotypes differed by mutations at only one or two positions whereas others were more divergent. Major findings were that one cpDNA haplotype predominated across much of Europe, that others closely related to it were geographically localized, and that one highly
distinct set of haplotypes occurred in Crimea. The widespread haplotype is probably an ancestral sequence for western Europe (from which other related haplotypes arose by recent mutations), and the distinctive haplotypes in Crimea probably register the former presence of a second Pleistocene refugium for this species in eastern Europe.
Vagile creatures can be genealogically structured, too
Many organisms have high dispersal potential, either as adults (for example winged insects, birds, or open-water marine fishes), as juveniles (for example marine invertebrates with long-lived pelagic larvae), or as gametes (for example plant species with wind-dispersed pollen). Even in such vagile species, however, historical barriers to dispersal can sometimes be insurmountable. For example, the Panamanian Isthmus has blocked inter-ocean gene flow in nearly all marine species since its origin 3 Ma. This is reflected today in the large number of marine fish, shrimp, sea turtle and other species whose populations on the Atlantic vs. Pacific side of the land bridge have proved to display quite deep historical separations in molecular surveys of mtDNA and other genes (Knowlton et al., 1993; Bermingham et al., 1997).
Sometimes, deep genetic splits distinguish extant populations that today do not appear to be sundered by obvious dispersal filters. A case-in-point involves a marine killifish (Fundulus heteroclitus) that inhabits the US Atlantic coastline, where a pronounced genealogical distinction between northern and southern populations is registered both in mtDNA and in a sequenced nuclear gene encoding lactate dehydrogenase (Bernardi et al., 1993). This example of aspect II concordance
may evidence a former presence of the species in two isolated Pleistocene refugia where genetic differentiation accumulated, followed by range expansions and secondary contact. This is not to say that natural selection has not also played a role in shaping this phylogeographic outcome. For example, natural selection in the past may have promoted some of the genetic divergence that took place in allopatry, and contemporary natural selection (from environmental gradients such as water temperature) may now inhibit haplotype exchanges between the northern and southern regions. A similar example involving aspect II of genealogical concordance, and for which similar kinds of explanations may apply, involves a copepod (Tigriopus californicus) distributed along the US Pacific coast (Burton & Lee, 1994).
Some organisms with high dispersal potential may show evident population genetic structure resulting from philopatry (behavioural fidelity to specific locations) rather than from physical barriers per se. Several cetacean species, for example, are matrifocal (socially organized around female kin), one expected genetic signature being a matrilineal population genetic structure. In mtDNA surveys of humpback whales (Megaptera novaeangliae), populations within each of three ocean basins did indeed prove to be subdivided into matrilineal stocks that probably arose from and are maintained by female faithfulness to particular migratory destinations (Baker et al., 1993). This site fidelity probably develops when a calf accompanies its mother on her annual migration between high-latitude and low-latitude waters.
Migratory birds are likewise highly vagile, but several surveyed species have proved to display significant phylogeographic structure on breeding grounds (and sometimes on migratory routes and in wintering areas also). In the dunlin shorebird (Calidris alpina), for example, five mtDNA clades correspond to regional breeding populations in Europe, eastern Siberia, central Siberia, Alaska and Canada (Wenink et al., 1996). This kind of population genetic architecture in a long-distance migrant is evidently the result of inter-generational fidelity to particular migratory circuits.
Not all species are phylogeographically subdivided
Although most species show significant phylogeographic structure, exceptions do exist, and these can be highly informative about broader phylogeographic principles. One
species that seems to lack salient phylogeographic partitions is the red-winged blackbird (Agelaius phoeniceus), one of the most abundant birds in North America. In a genetic survey of 127 specimens from across the continent, 34 distinct mtDNA haplotypes were detected, but the most common (and nearly ubiquitous) of these proved to be the core of a ‘starburst’ genealogy to which most of the other haplotypes (each typically rare) were independently linked (Ball et al., 1988).
Such starburst patterns are an expected signature for a species that may have expanded recently from a single geographic source; the common and widespread haplotype is probably the ancestral condition from which the rare haplotypes were recently derived by separate mutations. Thus, genealogical data from mtDNA suggest that, following the retreat of the most recent trans-continental glaciers (beginning about 18,000 years ago), red-winged blackbirds greatly expanded their range and population size as they recolonized the continent primarily from one Pleistocene refugium.
Another example of this general sort is provided by the American eel (Anguilla rostrata). This species is catadromous, meaning that individuals spend most of their lives in streams or lakes but then eventually migrate to the sea to spawn and die (the reverse of anadromous species such as salmon, which live in the sea but then migrate to spawn in streams). The spawning destination of American eels is the Sargasso Sea in the western tropical mid-Atlantic Ocean, where migrants from across eastern North America apparently congregate in what was long suspected to be a single panmictic (random-mating) event. The resulting larvae then eventually make their way back to freshwater habitats. Does this peculiar lifecycle mean that juvenile eels from throughout their vast freshwater range are all part of the same well-mixed gene pool? Phylogeographic data from mtDNA and nuclear genes are quite consistent with this possibility. Haplotypes and alleles are widely shared (and in similar frequencies) in surveyed eel populations from Maine to Louisiana (Avise et al., 1986).
For any such species that consists of a single well-mixed gene pool, coalescent theory can be applied to genealogical data to estimate that species’ long-term effective population size. The effective number (Ne) of individuals refers to the size of an idealized population that has the same genetic properties (such as mean coalescent times) as those observed in a real population. Usually, a long-term or evolutionary Ne is much smaller than N (the census size) for one or more of the following reasons: the number of breeding males may differ from the number of breeding females; natural populations typically fluctuate in size through time, and may even experience occasional bottlenecks; some individuals may leave many more progeny than others, creating a large fitness variance across families; selective sweeps may occur in which a positively selected mutation sweeps to fixation in a population or species, thereby in effect squeezing ancestry through fewer individuals than would otherwise be the case; or other factors such as periodic extinction and recolonization, which can make a species as a whole have a much lower Ne than might have been predicted had only the composite census sizes been available. The coalescent approach towards estimating evolutionary Ne involves estimating, from molecular data, a histogram of evolutionary times to shared ancestry for all pairs of assayed specimens, and then comparing that ‘mismatch distribution’ to coalescent expectations (under neutrality theory) for hypothetical populations of different effective sizes. For the eels, pairwise estimates of mtDNA sequence divergence were converted to coalescent times by assuming that the animals have a generation length of 10 years and that their mtDNA evolves at a standard pace (2% sequence divergence per million years). The resulting mismatch distribution proved to be close in magnitude and pattern to coalescent expectations for a single population of evolutionary effective size Ne = 5,500 females (Avise et al., 1988).
Qualitatively similar outcomes have been reported for several other species that seem to be genetically well mixed. Typically (as in the American eels), evolutionary effective population sizes are orders of magnitude smaller than currentday census population sizes, probably because each species has experienced population bottlenecks at one or more times in its recent evolution, and/or because of other demographic factors (mentioned above) that can likewise depress long-term effective population sizes below typical census levels.
Historical processes have also impacted biotic
communities
Several regional or landscape genetic analyses have compared phylogeographic outcomes across multiple co-distributed species (Bermingham & Moritz, 1998). A remarkable but rather consistent finding is that species with similar ranges or ecologies often tend to be genealogically structured in similar ways. This suggests that historical biogeographical forces are not merely species-idiosyncratic in their effects, but instead have also concordantly moulded the genetic architectures of particular regional biotas.
An example of aspect III of genealogical concordance is provided by genetic surveys of coastal faunas in the southeastern United States, where Gulf of Mexico populations in each of several species tend to be highly distinct genetically from most populations along the Atlantic Coast (Avise, 1992). Deep genetic distinctions between Atlantic and Gulf populations are not universal to all species surveyed (American eels provide a striking counter-example), but the general pattern does suggest that a significant fraction of the coastal biota has been genetically impacted in a rather concordant fashion. The phylogeographic factors responsible in this case are not entirely clear, but a working hypothesis is that ancestral populations in the Gulf and Atlantic were historically separated, perhaps during the Pleistocene, and accumulated genetic differences that today may be maintained by ecological obstacles (such as ocean currents or differences in water temperature) to interregional gene flow.
Concordant genealogical patterns for which the historical
explanations are somewhat clearer involve floras and faunas in Europe. Genetic surveys have provided strong evidence that three European regions not covered by Pleistocene ice masses – the Iberian Peninsula, the Italian Peninsula and the Balkan region – were primary refugia for numerous animal and plant species that in more recent times have recolonized the continent (Hewitt, 2000; Weiss & Ferrand, 2006). Phylogeographic footprints are evident today in the spatial arrangements within many species of distinct genealogical clades (sometimes termed intraspecific ‘phylogroups’) that trace back though various colonization routes to one or more of these Pleistocene refugia. In many cases, post-Pleistocene range expansions also brought such distinct phylogroups into secondary contact at suture zones, where hybridization often occurs. These suture zones are concentrated in specific areas (notably the Alps, central Europe and Scandinavia), apparently because of commonalities of ice-age refugia, histories of postglacial expansion and physical impediments to dispersal.
History can sometimes repeat itself
Another illustration of all four aspects of genealogical concordance involves freshwater fishes in the south-eastern United States. For each of several species surveyed for mtDNA and in some cases nuclear genes, populations in western rivers (draining into the Gulf of Mexico) are genetically distinct from populations in most eastern rivers (draining into the Atlantic Ocean) and peninsular Florida (Avise, 2000). Similar suture zones also exist where the two respective forms sometimes overlap and hybridize. These findings strongly imply the former presence of two separate refugia in which genetic differences accumulated and from which the subsequent range expansions occurred.
Although these phylogeographic patterns are generally concordant across multiple taxa, the apparent depths of the genealogical splits differ considerably from species to species. One possibility is simply that molecular evolutionary rates differ across these taxa, but a more intriguing hypothesis is that various extant species might have been historically sundered at different times. Across the 2 Myr of the Pleistocene, continental glaciers advanced and retreated cyclically, thereby repeatedly creating and dissolving particular refugia. Perhaps some modern fish populations in the eastern vs. western drainages ultimately trace back to the same refugial areas but at different times in this recurrent cycle. This would require that an ancestral population in one refugium went extinct during one or more of the cycles and later was replaced by colonizers from the other. The net result would be concordant spatial arrangements of gene trees but different temporal depths for their most ancient splits.
This possibility, although highly speculative, is also consistent
with a different type of biogeographical evidence: community structures. Several fish species in the south-eastern United States have ranges generally confined to either eastern (Atlantic) or western (Gulf) rivers, suggesting that populations formerly inhabiting one or the other of these areas became extinct (or perhaps diverged so much as to have been given separate species status). If so, this would help to explain not only the overall faunal patterns mentioned above, but also why these two biogeographical provinces are generally concordant with the two primary molecular phylogroups present within particular species.
Even a mitochondrial gene tree alone can give key biological insights
Although phylogeographic analyses ideally are based on nuclear as well as on mitochondrial data, a mtDNA genealogy
alone can suffice for some purposes because of its special relevance to female population demography (Avise, 1995). Imagine, for example, a species in which females are sedentary but males move large distances and mate at random with females from any site. Matrilines in this species would be dramatically structured, but nuclear genealogies would be geographically randomized. Despite the blatant discord between mtDNA and nuclear gene trees, the matrilineal structure itself would mean that each geographic location is essentially autonomous with regard to reproductive output, and this in turn could have major ramifications for population management or conservation.
To make the argument more concrete, consider the green sea turtle (Chelonia mydas). At various rookery sites scattered around the world’s tropical oceans, females lumber ashore to
lay their eggs. Otherwise, green turtles spend their lives at sea, foraging at locations that may be hundreds or thousands of kilometres from the nesting beaches, and thus necessitating Herculean reproductive migrations. Genetic surveys have revealed that most rookeries are clearly distinct from one another in mtDNA haplotypes (Bowen & Avise, 1996), indicating that nesting females have natal-homing tendencies (because if they did not, mtDNA haplotypes would be more thoroughly mixed among rookery sites). So, even if turtles from all rookeries mated randomly, and even if their nuclear genomes were thoroughly homogenized as a result (empirically they are not), the natal-homing propensity of females as documented in mtDNA would demonstrate a considerable degree of demographic autonomy for each rookery. This inference has conservation relevance: if any rookery were
extirpated by natural or human causes, its re-establishment by females hatched elsewhere would not be likely, at least in the short term. (Of course, departures from strict natal homing must also occur occasionally, or the species would not consist of multiple rookeries.)
Other contexts likewise arise in which a species’ matrilineal history per se is of interest. Green turtles again provide an example. One major rookery for this species is Ascension, a tiny volcanic island halfway between South America and Africa. Each nesting season, females, who otherwise have been feeding along Brazilian shorelines, embark on what will be a 4,400-km round-trip nesting odyssey to Ascension and back.
Why undertake this journey, which requires astonishing physiological endurance and navigational feats, when closer rookery sites are clearly available along the South American coast? One intriguing possibility raised long ago by Archie Carr (a pioneering sea turtle biologist) is that turtles began nesting on a proto-Ascension Island about 80 Ma, when South America and Africa were separated by only a narrow sea channel. Across the ensuing millennia, as the continents slowly drifted apart by plate tectonics, natal-homing females lengthened migratory circuit incrementally, by a few centimetres per generation.
This hypothesis, if true, requires an unbroken chain of matrilineal ancestry for Ascension nesters across 80 Myr, which in turn predicts that large differences in mtDNA sequence have accumulated between Ascension females and those nesting in South America or elsewhere. The Carr hypothesis was put to empirical test, and falsified (Bowen et al., 1989). The mtDNA haplotypes of Ascension females do differ from those of South American nesters, but the magnitude of sequence divergence is very small, indicating that the matrilines now inhabiting Ascension probably trace back to colonizers that arrived recently (within the last 100,000 years at most).
PROSPECTS FOR PHYLOGEOGRAPHY
Across the past three decades, phylogeographic perspectives (and the mitochondrial and other gene-tree systems from which these perspectives arose) have added many oft-unorthodox insights into evolutionary genetic processes (Avise, 2007). Table 1 briefly summarizes 21 such conceptual and empirical contributions. Phylogeography as a discipline has grown fantastically during the last 30 years, and shows no signs of slowing down. Indeed, I see ample opportunity for
expansion on at least three overlapping fronts.
Expand empirical analyses to multiple nuclear loci
Most phylogeographic analyses to date have focused primarily on cytoplasmic genomes (for several logically defensible reasons discussed earlier). The informational harvest from this relatively low-hanging fruit has been huge, but, looking forward, quantum advancements in the field could come from genealogical appraisals of the potentially juicier but harder-toreap nuclear genome. Overcoming the technical hurdles (such as how to rapidly isolate nuclear haplotypes from diploid
individuals) and the natural hurdles (especially how to deal
with intragenic recombination) may be daunting, but any researcher broadly successful in such endeavours will be rightly considered a scientific pioneer. The goal will be to recover gene genealogies from multiple unlinked nuclear loci in order to search for patterns of inter-locus genealogical concordance (or lack thereof) that will yield richer interpretations of the historical record for each species.
|
Table 1 Many unorthodox perspectives on evolution have been prompted by molecular discoveries about mtDNA and by the phylogeographic perspectives that those findings helped to motivate. For elaboration, see the current text and also Avise (2000, 2007).
1. Population hierarchy. Entire populations of mtDNA molecules inhabit somatic and germ cell lineages, thus adding an entirely new level to the population genetic hierarchy.
2. Germline bottlenecks. Homoplasmy typifies animal mtDNA, implying that population bottlenecks in germ cell lineages probably attend mitochondrial inheritance.
3. Conserved function yet rapid evolution. Rapid evolution of animal mtDNA challenges the idea that functional constraints necessarily set a low speed limit on sequence change.
4. Asexual transmission. Cytoplasmic genomes within sexual species normally exhibit clonal (uniparental, non-recombinational) genetic transmission.
5. Matrilineal histories. The rapid pace of mtDNA evolution, coupled with the molecule’s maternal inheritance, means that the intraspecific matrilineal histories can be recovered.
6. Individuals as operational taxonomic units. Because each animal has a specifiable mtDNA haplotype, each individual can be treated as an ‘operational taxonomic unit’ in genealogical appraisals.
7. Gene tree concept. In sexual organisms, mtDNA provides a prototype example of a non-anastomose or non-reticulate gene genealogy, or ‘gene tree’.
8. Gene tree concept extended. In principle, the multi-generation pedigree of any sexual species contains, in the nuclear genome, multitudinous nonisomorphic gene trees.
9. Gene trees vs. species trees. Gene trees for unlinked nuclear loci can and often do differ topologically from a composite species phylogeny; discordances between gene trees and species trees, owing to idiosyncratic lineage sorting, can also characterize extant taxa that separated anciently.
10. Genealogical topologies. With respect to particular gene trees, the phylogenetic status of any pair of sister species is itself likely to be evolutionarily dynamic, with a common time-course of events following population or species sundering being polyphyly fi paraphyly fi reciprocal monophyly.
11. Genealogical concordance. Because of the idiosyncratic nature of individual gene trees, deep splits at the population or species level can be recognized only by reference to any of several different aspects of ‘genealogical concordance’ (across loci, across species, or across different lines of evidence).
12. Genealogy and demography. Within any species, coalescent processes describe the connections between genealogy and population demography and show that genealogy and demography are thoroughly intertwined concepts.
13. Genealogy and gender. Key demographic and behavioural parameters often differ between males and females in ways that impact sex-specific genealogical structures.
14. Effective population size. Empirical gene-tree surveys, interpreted by coalescent theory, imply that most species have surprisingly small evolutionary effective population sizes.
15. Conspecific populations. Empirical data reveal that particular species are often subdivided into readily distinguished genealogical units (intraspecific ‘phylogroups’).
16. Phylogroup numbers. Empirically, the total number of highly distinctive intraspecific phylogroups per species has typically proved to be small (c. 1–6).
17. Spatial orientation. Intraspecific phylogroups are nearly always allopatric and typically spatially oriented in ways that seem to make good biogeographical sense.
18. Natural history. Genealogical data indicate that, in addition to historical biogeographical events, behaviours and natural histories of species also impact phylogeographic patterns.
19. Taxonomy and conservation. Phylogeographic data and concepts are highly germane to taxonomic judgments and also to conservation biology.
20. Phylogenetic reasoning. In general, and contrary to conventional wisdom, the language and concepts of phylogeny (i.e. genealogy) are highly relevant at the intraspecific level.
21. Microevolution, like macroevolution, is historical. In general, intraspecific evolution seldom produces the kinds of equilibrium outcomes (such as those derived under an island model of population structure) that were the mathematically tractable focus of much of traditional population genetic theory; instead, intraspecific genetic architectures are inherently historical (genealogical) and idiosyncratic to biogeographical context. The field of phylogeography explicitly recognizes and accommodates this fundamental reality.
|
Expand coalescent theory
Coalescent theory has developed primarily with a single-locus orientation, so considerable room exists for addressing interlocus variances in coalescent outcomes as a function of various genetic factors (such as the magnitude of linkage between loci) and of past and present population demographic parameters.
A meaningful multi-locus coalescent theory also will have to accommodate the wide variety of spatial population architectures that nature can potentially produce. Associated with these efforts will be the need to further integrate (align) the expectations of coalescent theory and other analytical methods (Excoffier, 2004) with empirical data into more coherent and intelligible wholes. The net goal should be a robust ‘statistical phylogeography’ (Knowles & Maddison, 2002; Templeton, 2004; Knowles, 2008) that reflects a realistic integration of population genetic and phylogenetic methods.
Extend phylogeographic analyses to additional taxa
Although many hundreds of empirical studies in phylogeography have been published, only a tiny fraction of the biological world has been examined from a phylogeographic perspective. Thus, future efforts will extend phylogeographic appraisals to many additional taxa. Much of this work will be motivated by a desire to understand the idiosyncratic genealogical histories of particular species that may be of special interest because of taxonomic uncertainties, conservation concerns, fascinating natural histories, or other biological motivations that may be species-specific. A major research focus will also be on comparative phylogeographic appraisals of multi-species regional biotas. Based on past experience (such as with various taxa in the south-eastern United States and in Europe), shared phylogeographic patterns (concordances) reflective of salient historical events will probably prove to characterize multiple species in many regional faunas and floras. Indeed, collaborative efforts are already underway to inventory phylogeographic patterns on several continents and in various subcontinental regions, the goal being to reconstruct major trends in the recent histories of vicariance and dispersal in each such corner of the world.
THE PLACE OF PHYLOGEOGRAPHY IN THE BIODIVERSITY SCIENCES
Before the initial ‘phylogeographic revolution’ of the late 1970s and 1980s, relatively little communication took place between biologists studying intraspecific (microevolutionary) genetic processes and those analysing supraspecific (macroevolutionary) genetic patterns. The former’s realm was population genetics, which dealt with changes in population allele frequencies resulting from mutation, genetic drift, gene flow, natural selection and sexual selection. The latter’s was phylogenetics and systematics, which dealt mostly with evolutionary relationships of species and higher taxa. The typical background of a population geneticist might be statistics, mathematics, ecology, or population demography, with perhaps little exposure to phylogenetic reasoning. The background of a systematist might involve museum or field work on a particular taxonomic group, with perhaps little exposure to population genetics. This state of affairs was both unfortunate and ironic, because population genetics and phylogenetic biology must in truth be parts of a temporal evolutionary continuum.
Because phylogeography tends to refer mostly to studies of conspecific organisms and closely related species, it could be considered a branch of population genetics. But it departs from classical population genetics by its special focus on genealogy (not just allele frequencies) as revealed by historical genetic records in particular pieces of DNA. In essence, intraspecific genealogy can be interpreted as phylogeny viewed up-close.
Because phylogeography deals with genealogy (micro-phylogeny), it also could be considered a branch of phylogenetic biology. But it departs from traditional phylogenetics by its special focus on population history and demography. A traditional sentiment among systematists was that phylogeny had no meaning at the intraspecific level because genetic lineages are reticulate or anastomose in interbreeding populations.
As we have seen, however, stretches of linked nucleotides (mtDNA and cpDNA are merely two examples) do have tree-like histories within a species, and the branching structures of these gene genealogies bear considerable analogy (albeit often on a different temporal scale) to phylogenetic trees summarizing historical relationships among reproductively isolated species and higher taxa. Furthermore, systematists seldom considered historical population demography as relevant in reconstructing species phylogenies, but in truth the historical demographies of conspecific organisms have an important and lasting impact on how hereditary lineages are apportioned among populations and species during the evolutionary process. Indeed, phylogeography and coalescent theory have made it abundantly clear that genealogy and historical population demography are intimately interconnected.
Figure 2 The general place of phylogeography, and some of its empirical and conceptual bridging functions, within the biodiversity sciences (modified from Avise, 2000).
Finally, phylogeography deals with the spatial and temporal dimensions of genealogy, so it also could be considered a branch of biogeography (perhaps its most natural home). But again it differs from traditional biogeography by its special focus on conspecific populations and on explicit genealogical information. It also differs from ecogeography (an important sub-branch of biogeography; Gaston et al., 2008) by its special focus on historical causation in addition to the selective forces and other ecological processes operating across more contemporary timeframes.
Through its novel genealogical perspectives and its obvious linkages to several different fields, phylogeography will continue to provide a powerful empirical and conceptual bridge between several major evolutionary disciplines that formerly had only limited communication (Fig. 2).
ACKNOWLEDGEMENTS
Recent work in the Avise laboratory has been supported by funds from the University of California at Irvine.
REFERENCES
Avise, J.C. (1992) Molecular population structure and the biogeographic history of a regional fauna: a case history with lessons for conservation biology. Oikos, 63, 62–76.
Avise, J.C. (1995) Mitochondrial DNA polymorphism and a connection between genetics and demography of relevance to conservation. Conservation Biology, 9, 686–690.
Avise, J.C. (2000) Phylogeography: the history and formation of species. Harvard University Press, Cambridge, MA.
Avise, J.C. (2007) Twenty-five key evolutionary insights from the phylogeographic revolution in population genetics.
Phylogeography of southern European refugia (ed. by S. Weiss and N. Ferrand), pp. 7–21. Springer, Dordrecht.
Avise, J.C. & Ball, R.M., Jr (1990) Principles of genealogical
concordance in species concepts and biological taxonomy.
Oxford Surveys in Evolutionary Biology, 7, 45–67.
Avise, J.C., Giblin-Davidson, C., Laerm, J., Patton, J.C. & Lansman, R.A. (1979) Mitochondrial DNA clones within and among geographic populations of the pocket gopher, Geomys pinetis. Proceedings of the National Academy of Sciences USA, 76, 6694–6698.
Avise, J.C., Helfman, G.S., Saunders, N.C. & Hales, L.S. (1986) Mitochondrial DNA differentiation in North Atlantic eels: population genetic consequences of an unusual life history pattern. Proceedings of the National Academy of Sciences USA, 83, 4350–4354.
Avise, J.C., Arnold, J., Ball, R.M., Jr, Bermingham, E., Lamb,
T., Neigel, J.E., Reeb, C.A. & Saunders, N.C. (1987) Intraspecific phylogeography: the mitochondrial DNA bridge
between population genetics and systematics. Annual Review of Ecology and Systematics, 18, 489–522.
Avise, J.C., Ball, R.M., Jr & Arnold, J. (1988) Current versus historical population sizes in vertebrate species with high gene flow: a comparison based on mitochondrial DNA lineages and inbreeding theory for neutral mutations.
Molecular Biology and Evolution, 5, 331–334.
Baker, C.S., Perry, A., Bannister, J.L., Weinrich, M.T., Abernethy, R.B., Calambokidis, J., Lien, J., Lambertsen, R.H., Urban Ramirez, J., Vasquez, O., Clapham, P.J., Alling, A., O’Brien, S.J. & Palumbi, S.R. (1993) Abundant mitochondrial DNA variation and world-wide population structure in humpback whales. Proceedings of the National Academy of Sciences USA, 90, 8239–8243.
Ball, R.M., Jr, James, F.C., Freeman, S., Bermingham, E. &
Avise, J.C. (1988) Phylogeographic population structure of
red-winged blackbirds assessed by mitochondrial DNA. Proceedings of the National Academy of Sciences USA, 85, 1558–1562.
Bermingham, E. & Moritz, C. (eds) (1998) Comparative phylogeography: concepts and applications. Molecular
Ecology, 7 (Special Issue), 367–545.
Bermingham, E., McCafferty, S.S. & Martin, A.P. (1997) Fish
biogeography and molecular clocks: perspectives from the
Panamanian Isthmus. Molecular systematics of fishes (ed. by T.D. Kocher and C.A. Stepien), pp. 113–128. Academic
Press, San Diego, CA.
Bernardi, G., Sordino, P. & Powers, D.A. (1993) Concordant mitochondrial and nuclear DNA phylogenies for populations of the teleost fish Fundulus heteroclitus. Proceedings of the National Academy of Sciences USA, 90, 9271–9274.
Bowen, B.W. & Avise, J.C. (1996) Conservation genetics of marine turtles. Conservation genetics: case histories from nature (ed. by J.C. Avise and J.L. Hamrick), pp. 190–237.
Chapman & Hall, New York. Bowen, B.W., Meylan, A.B. & Avise, J.C. (1989) An odyssey of the green sea turtle: Ascension Island revisited. Proceedings of the National Academy of Sciences USA, 86, 573–576.
Brown, W.M., George, M., Jr & Wilson, A.C. (1979) Rapid evolution of animal mitochondrial DNA. Proceedings of the
National Academy of Sciences USA, 76, 1967–1971.
Burton, R.S. & Lee, B.-N. (1994) Nuclear and mitochondrial gene genealogies and allozyme polymorphism across a major phylogeographic break in the copepod Tigriopus californicus. Proceedings of the National Academy of Sciences USA, 91, 5197–5201.
Dawkins, R. (1995) River out of Eden. Basic Books, New York.
Demesure, B., Comps, B. & Petit, R.J. (1996) Chloroplast DNA phylogeography of the common beech (Fagus sylvatica L.) in Europe. Evolution, 50, 2515–2520.
Excoffier, L. (ed.) (2004) Analytical methods in phylogeography. Molecular Ecology, 13 (Special Issue), 727–980.
Gaston, K.J., Chown, S.L. & Evans, K.L. (2008) Ecogeographical rules: elements of a synthesis. Journal of Biogeography, 35, 483–500.
Hare, M.P. (2001) Prospects for nuclear gene phylogeography. Trends in Ecology and Evolution, 16, 700–706.
Hewitt, G. (2000) The genetic legacy of the Quaternary ice
ages. Nature, 405, 907–913.
Hudson, R.R. (1990) Gene genealogies and the coalescent
process. Oxford Surveys in Evolutionary Biology, 7, 1–44.
Knowles, L.L. (2008) Statistical phylogeography: the use of coalescent approaches to infer evolutionary history.
Annual Review of Ecology, Evolution, and Systematics, 39,
in press.
Knowles, L.L. & Maddison, W.P. (2002) Statistical phylogeography. Molecular Ecology, 11, 2623–2635.
Knowlton, N., Weight, L.A., Solózano, L.A., Mills, D.K. &
Bermingham, E. (1993) Divergence of proteins, mitochondrial DNA, and reproductive compatibility across the Isthmus of Panama. Science, 260, 1629–1632.
Palmer, J.D. (1990) Contrasting modes and tempos of genome evolution in land plant organelles. Trends in Genetics, 6, 115–120.
Templeton, A.R. (2004) Statistical phylogeography: methods of evaluating and minimizing inference errors. Molecular Ecology, 13, 789–809.
Weiss, S. & Ferrand, N. (eds) (2006) The phylogeography of southern European refugia. Springer, Dordrecht
Wenink, P.W., Baker, A.J., Rösner, H.-U. & Tilanus, M.G.J.
(1996) Global mitochondrial DNA phylogeography of
holarctic breeding dunlins (Calidris alpina). Evolution, 50,
318–330.
BIOSKETCH
Research in the Avise laboratory centres on the behaviour,
natural history and evolution of animals as revealed by molecular markers. Concepts and theories of population genetics, phylogeography and speciation are integrated to address problems that are also often germane to conservation biology. Avise is the author of approximately 300 research articles and more than a dozen books on evolution and genetics.
Editor: Brett Riddle
This invited paper was occasioned by John Avise’s recent receipt of the Alfred Russel Wallace Award, from the International Biogeography Society, for career contributions to biogeography.
|
Mencionamos aqui alguns exemplos do caminho actual desta nova e fascinante disciplina da filogeografia relacionados às questões de refúgios glaciares e evolutivos da Serra da Estrela e da Península Ibérica. Um bom tratado das espécies refugiais da Península Ibérica é o Capítulo V: “Refugia within refugia: patterns of phylogeographic concordance in the Iberian Peninsula” de Africa Gómez e David H. Lunt que se encontra no livro de Steven Weiss and Nuno Ferrand (eds.), Phylogeography of Southern European Refugia 155-188. 2006. Um resume mais recente sobre espécies refugiais mediterrânicas e não-mediterrânicas é o artigo. “Extra-Mediterranean refugia: The rule and not the exception?” na revista “Frontiers in Zoology” de 2012. Vamo-nós orientar nestes artigos, mas juntaremos informação adicional sobre a espécie Galemys pyrenaicus, recentemente publicado.
No seguinte vamos comparar resultados de um estudo morfológico (2006) sobre Galemys pyrenaicus com resultados da análise genética (filogeográfica) sobre esta espécie (2013).

Veja à seguir: 13. The Northern Serras of Portugal (Serra da Estrela (Aspectos glaciários - d4))
[1] Willi Hennig: Phylogenetic Systematics. University of Illinois Press, 1999. (first edit.1966).
[2] Gareth Nelson and Norman Platnick: Systematics and Biogeography. Cladistics and Vicariance. Columbia University Press. New York 1981.
[3] Wakeley, J. 2008. Coalescent Theory: An Introduction. Roberts & Company Publishers, Greenwood Village, Colorado.
[4] John Wakeley (2003).Inferences about the structure and history of populations: coalescents and intraspecific phylogeography.
[5] Knowles LL. Statistical phylogeography. Annu. Rev. Ecol. Evol. Syst. 2009. 40:593–612
[6] Knowles LL, Maddison WP. 2002. Statistical phylogeography. Mol. Ecol. 11:2623–35
[7] ALAN R. TEMPLETON. Nested clade analyses of phylogeographic data: testing hypotheses about gene flow and population history. Molecular Ecology (1998) 7, 381–397
[8] ALAN R. TEMPLETON. Statistical phylogeography: methods of evaluating and minimizing inference errors. Molecular Ecology (2004) 13, 789–809
[9] Modelos de coalescência com seleção (veja p.e. Wakeley, 2010. Natural Selection and Coalescent Theory)
[10] Erik W. Bloomquist, Philippe Lemey and Marc A. Suchard.Three roads diverged? Routes to phylogeographic inference. Trends in Ecology and Evolution Vol.25 No.11
[11] Wakeley, J. 2010. Natural selection and coalescent theory. Pp. 119-149 in Evolution since Darwin: The First 150 Years (Michael A. Bell, Douglas J. Futuyma, Walter F. Eanes, and Jeffrey S. Levinton eds.), Sinauer and Associates, Sunderland, Massachusetts.
[12] Chapter 1 From: L. Lacey Knowles e Laura S. Kubatko: Estimating Species Trees: Practical and Theoretical Aspects. Edited by L. Lacey Knowles and Laura S. Kubatko. 2010 Wiley-Blackwell
[13] P. A. UNDERHILL, G. PASSARINO, A. A. LIN, P. SHEN, M. MIRAZÓN LAHR, R. A. FOLEY, P. J. OEFNER and L. L. CAVALLI-SFORZA: "The phylogeography of Y chromosome binary haplotypes and the origins of modern human populations. Ann. Hum. Genet. (2001), 65, 43-62
[14] Journal of Biogeography (J. Biogeogr.) (2009) 36, 3–15 (Download)
.
Sem comentários:
Enviar um comentário